Hepatocellular carcinoma. Diagnosis and treatment. Third edition. Brian I. Carr (editor) (2016)
Введение
Nonalcoholic fatty liver disease (NAFLD) describes a spectrum of liver diseases with pathology resembling liver damage induced by alcohol abuse but occurs in individuals who consume little or no alcohol. Histologically, the scope of NAFLD ranges from instances of simple fat accumulation in the liver to nonalcoholic fatty liver (NAFL) with isolated steatosis and mild nonspecific inflammation, to nonalcoholic steatohepatitis (NASH) [1–3]. NAFL is largely considered to be a benign condition whereas NASH is considered the more progressive subtype of NAFLD often characterized by diffuse hepatocellular injury which can progress to show evidence of necroinflammation, cirrhosis, and fibrosis and in some instances advance to hepatocellular carcinoma (HCC). NAFLD represents approximately 47% of chronic liver diseases in the US surpassing hepatitis B, hepatitis C, and alcoholic liver disease as the fastest growing cause of chronic liver disease in adults [4]. NASH-associated cirrhosis is the third most common cause of death in NAFLD patients and is predicted to surpass alcoholic liver disease and hepatitis C virus (HCV) to become the leading indication for liver transplantation in the U.S. over the next decade [5].
Инцидент/превалирование/факторы риска
NAFLD was first described in 1980 and has since become the most common cause of chronic liver disease worldwide. Few studies include long-term follow up of NAFLD patients. Thus the exact natural history of NAFLD is dif?cult to ascertain. The global prevalence of NAFLD is however rapidly increasing over time and is currently assumed to range from 20 to 45% in Western countries and 5–18% in Asia depending on the studied population and method of diagnosis [5–8]. In the United States, NAFLD is thought to affect approximately 34% of adults and 20% of children with an incidence running in parallel to the increased incidence of obesity and diabetes [7, 9, 10].
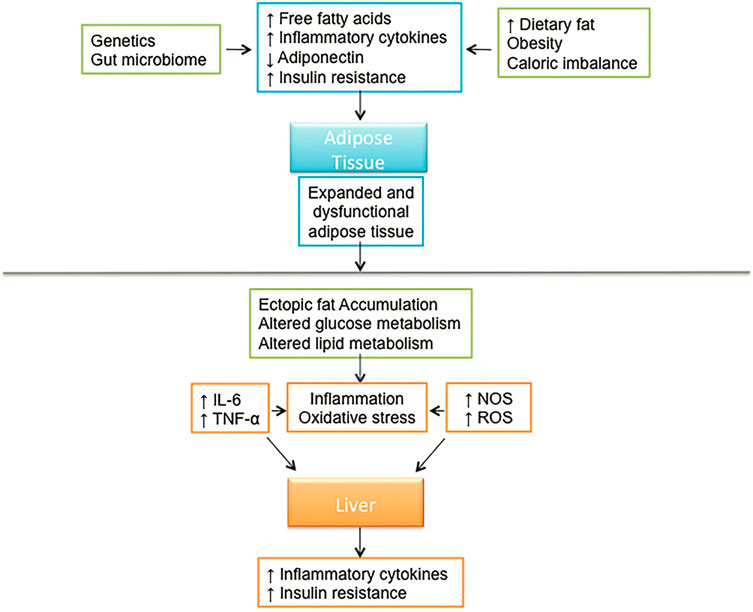
Фиг. 18.1. Системные и печень специфические механизмы, вовлеченные в патофизиологию NAFLD
The global incidence of NASH is estimated to be between 3 and 5%. Few noninvasive modalities exist which can differentiate NAFLD from NASH. However, population-based studies surveying levels of aminotransferases indicate that NASH affects between 6 and 8% of adults in the US [11]. A recent study suggests that the frequency of NASH varies significantly with ethnicity with significantly higher incidence in Hispanics (58.3%) compared to Caucasians (44.4%) and African Americans (35.1%) [7]. A close link has been identified between the metabolic syndrome and NAFLD. The metabolic syndrome can be defined by the presence of three or more of the following conditions: visceral obesity, hypertension, type 2 diabetes or elevated fasting plasma glucose, or dyslipidemia including hypertriglyceridemia or low high-density lipoprotein levels. Thus many consider NAFLD to be the hepatic component of the metabolic syndrome (Fig. 18.1).
Ожирение
The worldwide incidence of obesity has been rapidly progressing and is now described by the World Health Organization as a global epidemic. Recent studies suggest that there are 1.6 billion overweight and 500 million obese adults globally [12]. The prevalence of NAFLD is increased up to 80–90% of obese adults and 60% in hyperlipidemic adults [1, 13]. Approximately 69% of adults and 32% of children in the United States are currently considered overweight or obese. This translates to close to 100 million possible cases of NAFLD in the US alone [14, 15]. Recently, a prospective study reported improved clinical, metabolic, and biological outcomes in patients one year after bariatric surgery [16]. Following bariatric surgery, NASH had disappeared in 85.4% of the study group suggesting that the deleterious metabolic effects of NASH may in fact be reversible [16].
Резистентность к инсулину
Insulin resistance has been identified with an astounding frequency in individuals with NAFLD and is now thought to play an integral role in its pathogenesis. There appears to be a direct correlation between the degree of insulin resistance and the severity of NAFLD in patients and higher serum insulin levels are found in patients with NASH and fatty liver [17–19]. Furthermore, insulin resistance is consistently found in subjects with NAFL or NASH, even in the absence of diabetes [20]. Similar levels of insulin resistance have been observed in both overweight and lean patients with fatty liver suggesting that insulin resistance, not simply excess body fat, is essential to the pathogenesis of fatty liver disease [19].
The primary functions of adipose tissue are to store lipids, which can be burned to meet the energy needs of the body and to protect from excesses in circulating glucose by storing triglycerides produced by the liver from sugars. Ectopic fat accumulation describes the scenario wherein lipid accumulation has occurred in a site other than adipose tissue such as the liver, pancreas, or other organs not designed to accommodate excessive lipids loads [21, 22].
The two primary issues that induce ectopic fat accumulation are (1) an excess in energy intake as compared to expenditure, and (2) defects in mechanisms that control the proper shuttling of excess energy as lipids to adipose tissue. NAFLD is an example of ectopic fat accumulation. Hepatic lipid accumulation creates an insult to the liver which induces increased secretion of hepatokines, increased gluconeogenesis, decreased glycogen synthesis, and inhibition of insulin signaling [21, 23]. Excess fatty acids not only induce hepatic insulin resistance but also impair insulin clearance [24, 25].
Диабет
Type 2 diabetes (T2DM) and NAFLD are closely associated and NAFLD incidence is elevated in 69% of patients with type 2 diabetes mellitus. Studies show that type 2 diabetics have a 2to 4-fold increase in serious liver disease and are at increased risk of mortality from cirrhosis, and hepatocellular carcinoma [26–30]. Family history of diabetes and or insulin resistance also increases the risk of cirrhosis and fibrosis in diabetics and nondiabetic NAFLD/NASH patients alike [31, 32]. It has been documented that sustained elevation of plasma FFA levels over time can impair insulin secretion in lean, nondiabetic subjects who are genetically predisposed to T2DM [25]. A convincing body of evidence exists in support of the link between NAFLD and T2DM. Most estimates of T2DM in NAFLD have been based largely on medical history or the less sensitive plasma fasting glucose or A1c levels. Therefore, there is a need for well-controlled long-term prospective studies on the natural history of NAFLD in T2DM using more accurate methods of analysis.
There are also data suggesting that hypothyroidism, hypopituitarism, hypogonadism, sleep apnea, and polycystic ovary syndrome independent of obesity are important risk factors for the incidence of NAFLD [33–41]. Further investigation is warranted to determine if each of these factors truly influence the natural history of NAFLD or exist simply as comorbidities.
Патофизиология
Прогрессирование болезни
Fatty liver (steatosis) is the more common subtype of the fatty liver diseases and has long been considered as benign. NASH, seen in 10–25% of NAFLD cases, has been considered the more progressive disease state. Recent findings have prompted a shift in this paradigm. Where previous studies reported that NAFL may be benign, with little to no risk for progression to a more advanced disease, more recent studies provide evidence that progressive fibrosis can develop in both NASH and NAFL patients [42, 43]. In addition, it must be considered that NAFL can progress to NASH with fibrosis indicating that NAFL must also be considered a progressive disease [43]. Estimating the true incidence of NAFLD including NASH has been extremely challenging for many reasons including variability within study groups and lack of accurate noninvasive diagnostic techniques. Tracking the progression from one NAFL to NASH remains a challenge and the factors which may potentially cause progression from NAFL to NASH are still largely unknown.
The progression from non alcoholic steatohepatitis to cirrhosis and advanced fibrosis is a process that has been frequently studied [44–46]. NASH is a complex disease characterized by hepatocyte ballooning, macrovesicular steatosis, inflammation, and pericellular fibrosis. 15–20% of NASH cases progress to cirrhosis and the 5-year incidence of HCC in individuals with cirrhotic NASH is approximately 11% [47–49]. Hepatic fibrosis develops in 40–50% of patients with NASH [27]. The presence of advanced hepatic fibrosis is a key contributor to the development of HCC and a key predictor of all-cause and disease-specific mortality in NASH patients [50, 51]. Therefore, the presence of NASH may in and of itself be considered a risk factor for hepatocellular carcinoma. Recent studies indicate that the metabolic impact of obesity in nonalcoholic fatty liver disease may vary widely even among patients with a similar body mass index (BMI) [52, 53]. Therefore when describing the pathogenesis of NAFLD, it is important to consider that numerous factors contribute to its onset and progression. Based on current research, the disease can be attributed to any combination of genetic, dietary, inflammatory, and environmental factors (Fig. 18.2), which will be
urther discussed here.
Молекулярные механизмы
Вклад резистентности к инсулину
There appears to be a direct correlation between the degree of insulin resistance and the severity of NAFLD. It is now clear that adipose tissue dysfunction and inflammation play an integral role in the insulin resistance associated with NAFLD pathogenesis. Adipocytes protect the body from excess energy supply and excess ectopic triglyceride accumulation by activation of several inflammatory pathways. The activation and in?ltration of adipose tissue macrophages incite adipocyte dysfunction, adipose tissue insulin resistance, release of excess free fatty acids into the circulation, and ectopic fat deposition [54, 55]. There are two distinct classes of macrophages, which include: The “classically activated” (M1) macrophages and the “alternatively activated” (M2) macrophages. M1 macrophage activation is an essential component of humoral immunity stimulated by microbial products, and proinflammatory cytokines (ex. IFNc, TNF a). Upon activation, M1 macrophages secrete large amounts of proinflammatory factors including nitric oxide (NO) and reactive oxygen species intermediate (ROI) in addition to numerous proinflammatory cytokines (TNF-α, IL-1, IL-6, IL-12) [56]. By contrast, alternative/M2 macrophage activation is characterized by little to no secretion of proinflammatory cytokines, increased secretion of antiinflammatory cytokines, and enhanced scavenging of cellular debris [56, 57]. Stimulated by the presence of IL-4, 10 and 13, the M2 macrophage response is often associated with tissue remodeling and repair. Obesity generates a state of low grade inflammation and adipose tissue macrophage in?ltration often associated with insulin resistance [58, 59]. A systemic increase in the number of M1 relative to M2 macrophages is characteristic of human obesity and animals fed a high-fat diet [54, 57].
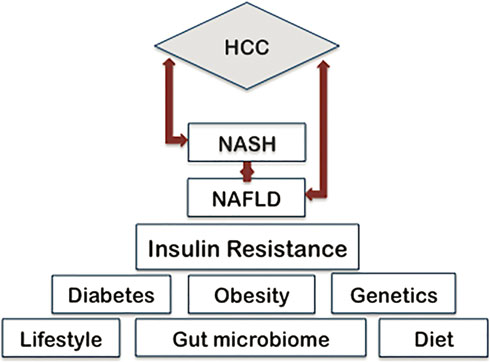
Фиг. 18.2. Прекурсоры и модификаторы, которые способствуют началу NAFLD и ее прогрессированию в NASH и HCС
Insulin resistance develops when macrophages invade visceral adipose tissue stimulating an inflammatory cascade that includes adipokine secretion [60]. Adiponectin and leptin are adipokines that decrease insulin resistance, while TNF-α, IL-6 and resistin, enhance insulin resistance. Reduced levels of adiponectin and elevated TNF-α and IL-6 are often synonymous with the NAFLD phenotype. Factors implicated in the initial genesis of adipose tissue inflammation, include relative ischemia and production of the hypoxia inducible factor-1, specific gut microflora, and microflora-dependent inflammatory responses and hormones such as leptin [60]. The combination of high circulating insulin levels and high plasma FFAs stimulates hepatic sterol regulatory element binding protein 1c (SREBP-1c) which in turn induces hepatic lipogenesis and oversecretion of very-low-density lipoprotein (VLDL) [24, 25, 61]. Increased lipid synthesis results in increased production of intermediates. By-products of this process include di-acylglycerols (DAG), di-palmitoyl phosphatic acid (Di-P PA), and ceramides [62–65]. DAG in particular is a known contributor to hepatic insulin resistance and is also involved in promoting hepatic inflammation [66, 67]. Elevated hepatic VLDL secretion lowers high-density lipoprotein levels and increases intrahepatic triglyceride accumulation [25, 61, 65]. Together these factors contribute to both chronic liver inflammation and hepatic insulin resistance. Several signaling pathways are known to be involved in this response. The c-Jun N-terminal kinase/activator protein 1, cyclic adenosine monophosphate responsive element binding protein H (CREB-H), the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway, and the nuclear factor jB (NFjB) pathways have been implicated in this process [68, 69]. These pathways are activated in response to elevated levels of fatty acids and lipid by-products. Excess in energy intake as compared to expenditure, as observed in obesity, may expose cells to toxic lipids, thereby activating cellular stress pathways. In addition, saturated fatty acids are known to disrupt endoplasmic reticulum (ER) homeostasis inducing ER stress and apoptosis in hepatocytes [70, 71]. This type of cellular stress originates from the accumulation of unfolded or misfolded proteins in the ER and often prompts an adaptive response including activation of the aforementioned pathways ultimately resulting in the release of reactive oxygen species and proinflammatory cytokines such as TNFa and IL-6 [54, 57, 60]. The metabolic consequence of this state is recognized as insulin resistance. Taken together it is clear that in addition to the hepatic milieu there is also a systemic syndrome. As such, the adipose tissue dysfunction and subsequent adipose tissue macrophage activation precede Kupffer cell activation.
Вклад микрофлоры кишечника
Several studies have provided evidence suggesting that dysbiosis of the gut microbiota may play a significant role in regulating intrahepatic metabolic and inflammatory pathways that contribute to the development and progression of NAFLD. The mechanisms responsible for this process are not completely understood but the increased intestinal absorption of multiple bacterial products, such as short-chain fatty acids, lipopolysaccharide (LPS), and endotoxins are thought to be involved [72]. Studies examining fecal microbiota in NAFLD and NASH patients have yielded interesting results. These studies reveal that the microbiota of patients with NAFLD or NASH have a lower proportion of members of the Ruminococcaceae family than healthy subjects [73–75]. Studies have shown that NASH patients have a higher prevalence of small intestinal bacterial overgrowth with elevated expression of TLR-4 and release of IL-8 [76]. A link between the percentage of gram-negative bacteroidetes and the presence of NASH has also been identified [77]. Furthermore, an increase in the abundance of alcohol-producing bacteria has been observed in NASH patients suggesting that these strains in particular may play a role in NASH pathogenesis [75]. Intestinal permeability and bacterial overgrowth correlate with severity of steatosis, but not fibrosis or hepatic inflammation. However, sustained exposure to these inflammatory mediators does promote the generation of various profibrogenic and apoptotic factors [78].
Changes in bacterial metabolites have been associated with obesity, and fatty liver disease. Notably, deficiency in the metabolite choline has been implicated in the pathogenesis of NAFLD and NASH. Diets high in fat and cholesterol promote the formation of intestinal microbiota that converts dietary choline into methylamines [79]. This process results in reduced circulating plasma levels of phosphatidylcholine. Phosphatidylcholine is required for assembly and secretion of VLDL and without it an accumulation of triglycerides is inevitably observed in hepatocytes [79]. Many of the complications of cirrhosis such as hepatic encephalopathy and infections have been linked to dysbiosis of the intestinal microbiome. Increased levels of endotoxin, systemic inflammation, and production of bacterial by-products such as ammonia contribute to pathogenesis. The products of hepatocyte injury and the cytokine milieu combine with systemic factors to promote inflammation within the liver creating a clear progression from fatty liver disease to steatohepatitis. The identification of pathogenic pathways linking the status of the gut to liver function has been eye opening. It is believed that these pathways are driven by dietary changes that could possibly induce gut dysbiosis potentiating hepatic inflammation and ultimately promoting hepatocarcinogenesis [77, 80, 81].
Вклад генетических факторов
The progression from NASH to end-stage liver disease, i.e., cirrhosis, and HCC is relatively infrequent. This suggests involvement of genetics factors influencing variables such as; hepatic innate immune function, lipid metabolism, extracellular matrix architecture, and cellular transformation resulting in the onset and progression of liver disease. Genome-wide association (GWAS) and candidate-gene studies have provided invaluable insights into the genetic contribution to NAFLD pathogenesis. The patatin like phospholipase 3 (PNPLA3) or adiponutrin gene was the first bona fide NAFLD-related gene to be identified using such methods [82, 83]. Individuals harboring the rs738409 C > G single-nucleotide polymorphism (SNP), encoding the Ile 148Met variant protein of PNPLA3 more frequently develop NASH [82]. The rs7384 mutation of PNPLA3 is not only associated with NASH, but also with the severity of necroinflammatory changes independent of metabolic factors and fibrosis [84, 85]. Subsequently, carriers of this mutation are at a threefold higher risk for NASH and 12-fold greater risk of HCC in comparison to noncarriers [86, 87]. The mechanisms underlying the role of PNPLA3 in liver disease are not well understood however the expression of the rs738409 variant is thought to interfere with lipoprotein export shifting the balance in favor of lipogenic activity over lipase activity, leading to hepatic fat accumulation [88–90]. Modifications of PNPLA3 remain the most verified genetic factor in the progression of NAFLD however the contributions of other genetic factors have been described.
Of note, a multi-ancestry, population-based exome-wide association study recently identified a nonsynonymous SNP in the transmembrane 6 superfamily member 2 (TM6SF2) gene producing a glutamate to lysine amino acid substitution at residue 167 (Glu167Lys) [91]. The TM6SF2 rs58542926 SNP (c.449 C > T, p.Glu167Lys) SNP is associated with increased hepatic triglyceride content and is highly conserved across mammals [91]. The TM6SF2 variant encoding p.Glu167Lys results in lowering of the levels of low-density lipoprotein cholesterol (LDL-C), triglycerides, and alkaline phosphatase in 3 independent populations. Carriage of the TM6SF2 minor allele is associated with NAFLD in general and advanced hepatic fibrosis/cirrhosis in particular and thus with increased risk of progression to NAFLD–HCC [92]. Most convincing perhaps was the reported gene-dosage effect, wherein the incidence of NAFLD increased with the number of minor alleles possessed [92]. The fact that hepatic triglyceride accumulation has not been directly linked to hepatotoxicity indicates that more research is required to determine the exact mechanism through which TM6SF2 drives NAFLD-associated hepatic fibrosis.
Диагноз
Non alcoholic fatty liver disease is largely asymptomatic particularly in its early stages. In some instances, patients report nonspecific symptoms such as fatigue and fewer still, report pain in the right upper quadrant. Currently, there are no defined symptoms of NAFLD or physical examination findings which clearly indicate the presence of the disease [93]. Abnormal liver function tests or incidental observations in patients undergoing thoracic and abdominal imaging for reasons other than liver symptoms, often lead to the diagnosis of NAFLD. Three distinct parameters are important for the diagnosis of NAFLD. These factors include evidence of hepatic steatosis by imaging or histology and the absence of competing etiologies for hepatic steatosis or significant alcohol consumption [13]. There are some clinical indicators that have been associated with NAFLD and associated disease. For instance, acanthosis nigricans resulting from insulin resistance is often associated with advanced disease, and the presence of a dorso-cervical hump has been linked to nonalcoholic steatohepatitis in some patients [93, 94]. Clinical manifestations including palmar erythema, spider angiomata, gynecomastia, or prominent upper abdominal veins may also be observed in patients following the onset of cirrhosis [93]. Cirrhosis is described as a progressive disease which starts with an initial asymptomatic or compensated phase and progresses to a more advanced decompensated phase marked by portal hypertension and liver dysfunction. Progression from compensated cirrhosis to decompensated cirrhosis is associated with a host of pathologies, such as ascites, jaundice, splenomegaly, and asterixis [93].
Биопсия печени
Liver biopsy remains the gold standard for identifying patients with NAFLD as it provides a definitive assessment of hepatic steatosis, hepatocellular injury, inflammation, and fibrosis. Numerous limitations are associated with biopsy including patient discomfort, procedure-related complications, sample variability, and observer variability [95]. Despite these limitations, liver biopsy remains the most consistent method of diagnosing and staging NASH. Identification of nonalcoholic steatohepatitis on an initial liver biopsy is a warning sign for the development of liver fibrosis [96]. The progression of liver fibrosis is a key predictor of all-cause and disease-specific mortality in NASH patients [51, 97]. Therefore, early diagnosis of nonalcoholic steatohepatitis and cirrhosis is essential from a treatment and management standpoint. Despite its clear utility, performing liver biopsy on every patient suspected of NAFLD would be impractical and it is thus essential to identify accurate and specific noninvasive methods to diagnose NASH. Several methods currently in use are described in the following section.
Трансаминазы
While mildly elevated transaminases [alanine aminotransferase (ALT) > aspartate transaminase (AST)] and/or gamma-glutamyltransferase (GGT) may be observed in some patients with NAFLD, over 50% of patients with advanced disease have normal liver enzyme levels [98, 99]. Additionally, ALT is an unreliable predictor of both steatosis and fibrosis in individual patients [98, 100].
Imaging
Recent innovations in imaging technology have shown potential to change how we both diagnose and monitor liver fat content. Ultrasound is an example of a low-cost, low-risk, and widely available diagnostic tool that may be utilized for qualitative assessment of hepatic disease. In the past, ultrasound was associated with numerous diagnostic limitations including an inability to distinguish NASH from NAFL and poor sensitivity for steatosis below 30% [101]. A newer quantitative ultrasound technology (QUS) has recently been developed to better characterize tissue microstructure by measuring fundamental acoustic parameters. Improvements on the previous ultrasound technique include the ability to more accurately measure liver fat even in the morbidly obese, and to the ability to identify the presence of steatohepatitis [102]. Potential issues are that results are operator dependent and interpreted qualitatively, therefore open to variability and subjectivity. With continued validation, this method shows promise as a noninvasive method to quantify hepatic steatosis. More studies are required to determine the efficacy of this method in assessing advanced liver disease. Another imaging modality that can be used to detect hepatic fat is magnetic resonance imaging (MRI), including magnetic resonance spectroscopy. Recent data suggest that magnetic resonance imaging and MRS may be a superior to histological evaluation in assessing longitudinal changes in liver fat content. This method detects the presence of hepatic fat greater than 5.56% with close to 100% accuracy [103]. And numerous studies evaluating the diagnostic performance of magnetic resonance MRI modalities for assessing hepatic steatosis and tracking effects of treatments in patients with NAFLD have shown great promise [104, 105]. Unfortunately, though this is both a sensitive and specific method of quantifying liver fat and steatosis, it is also expensive and not widely available. Efforts to increase the availability of MRI modalities will no doubt move us closer to the development of noninvasive determination of NAFLD that identifies the population at risk of worse outcomes and disease progression, tracks disease progression, and assess response to therapy.
Предиктивные модели
Several low-cost, noninvasive predictive panels have been developed for the assessment of fibrosis in chronic liver disease. The NAFLD fibrosis (NFS) and fibrosis score 4
(FIB-4) stand out as the most commonly used [106–108].
The FIB-4 score scoring system was originally developed as a predictive measure for advanced fibrosis in HIV patients also infected with hepatitis C [109]. Using this method age, AST, platelet counts, and ALT are evaluated and assessed as predictors of fibrosis. The NAFLD fibrosis score is considered the most validated and best performing predictive panel for evaluation of liver-related outcomes. When calculating the NFS, metabolic risk factors such as age, body mass index, and fasting glucose are evaluated alongside readily available clinical data including platelet count, albumin level, and the ratio of AST to ALT. Thus, NFS may offer a more comprehensive evaluation to identify patients at risk for severe disease. Using the NFS model, advanced fibrosis can be excluded in patients with a score below the low cut-off score of -1.455 (with 75% sensitivity and 58% specificity). Conversely, an NFS above 0.676 is an indicator of the presence of advanced fibrosis (with 33% sensitivity and 98% specificity) [110–112]. Thus, NFS may be utilized as a low-cost, noninvasive panel to aid in the identification of patients with liver disease who may benefit most from liver biopsy.
Биомаркеры NAFLD
In recent years, a number of biomarkers have been identified which are associated with NASH, such as cytokeratin-18 (CK-18) and terminal peptide of procollagen III (PIIINP). However, no single broadly validated biomarker has been found which can accurately and consistently diagnose NASH.
MicroRNAs (miRNA) are known to play an essential role in a variety of biological processes and have also been implicated in the progression of NAFLD [113]. A number of studies suggest that NAFLD has a distinguishing circulating miRNA profile that may be exploited for diagnostic purposes. MicroRNA-122 is perhaps the most wellcharacterized liver-associated miRNA as it is the most abundant miRNA found in the liver [114]. Closely linked to metabolic homeostasis, miR-122 has been shown to indirectly modulate the expression of genes involved in hepatic cholesterol and lipid metabolism [115–118]. Studies indicate that serum levels of miR-122 along with miRNAs 192, 375, and 19 were significantly elevated in patients with NAFLD as compared to healthy controls [119–122]. Furthermore, serum levels of miR-122 were shown to successfully distinguish NASH from simple steatosis and to identify liver fibrosis [122]. Based on these studies, circulating miR-122 might be useful as a biomarker for diagnosing fatty liver disease and monitoring the progression of histological changes, during therapeutic intervention.
Лечение
Изменение образа жизни
Питание
Individuals with NAFLD often have a diet high in saturated fat and cholesterol and may partake in overconsumption leading to energy imbalance and an overweight or obese phenotype. Studies indicate that even relatively moderate weight loss (as low as 10% of body weight) can improve hepatic insulin resistance and significantly reduce liver fat accumulation [123, 124]. In addition, massive weight loss following bariatric surgery can induce the disappearance of NASH and the partial reversal of cirrhosis in the liver [16, 125, 126]. Thus, it is clear that dietary intervention is an important component in treating NAFLD patients. Caloric restriction drives weight loss, visceral adiposity, subcutaneous fat, and liver fat reduction. Thus, reducing calories appears to be the most significant component of dietary intervention [127]. Also of importance to NAFLD progression, is the quality of dietary fat, as evidenced by the beneficial effects of mono and polyunsaturated fatty acids on fatty liver disease [128, 129]. As such, the Mediterranean diet which is rich in mono and polyunsaturated fatty acids has proven effective in reducing liver fat and improving hepatic insulin sensitivity even in the absence of significant weight loss [130–132].
Физическая нагрузка
It is not entirely clear if exercise exerts independent benefits in patients with NAFLD. The benefits of exercise in improvement of cardiovascular heath and reduction of the risks of the metabolic syndrome are, however, widely known. Fitness affects the response to calorie reduction; thus, improvement of cardiorespiratory ?tness may reduce liver fat with diet-induced weight loss [133]. Resistance exercise has been shown to reduce liver fat, improve insulin sensitivity, and promote fatty acid oxidation in NAFLD patients [134]. Improvement of overall ?tness may improve the resolution of NAFLD. The intensity and frequency of exercise required to realize improvements in NAFLD is poorly defined; thus, no specific recommendations can be made in this area.
Фармакологические агенты
Сенсибилизаторы инсулина
Insulin resistance is nearly universal in NAFLD patients and plays an important role in its pathogenesis by inducing peripheral lipolysis, de novo lipogenesis, and ectopic lipid accumulation. Hence, insulin sensitizers make for an attractive target for the treatment of NAFLD. Pioglitazone is the most well-studied pharmacological agent used for treatment of NASH and belongs to the class of drugs known as thiazolidinediones (TZDs). TZDs upregulate adiponectin, promote differentiation of insulin-sensitive adipocytes, enhance fatty acid uptake in adipose tissue, shuttle nonesteri?ed free fatty acids toward adipocytes, and reduce ectopic fat accumulation. Treatment with pioglitazone has been shown to resolve steatohepatitis with the improvement of all individual histological features except for fibrosis. Glitazones use has been associated with a number of side effects, particularly weight gain, which is not always reversible upon discontinuation. Pioglitizone has also been associated with postmenopausal bone loss and instances of congestive heart failure [135, 136].
Гепатопротекторные агенты
Витамин E
Vitamin E is a fat-soluble compound that is present in the phospholipid bilayer of cell membranes, the rationale for investigating the use of vitamin E in a NASH patient is based on the role of oxidative stress in NASH progression. Vitamin E is an antioxidant that prevents liver injury by protecting against free radicals and mitochondrial toxicity [137]. Vitamin E has shown moderate efficacy in improving inflammation and ballooning in NASH patients [138]. While generally considered benign, there have been reports of side effects with long-term use of vitamin E which include increased risks of prostate cancer and hemorrhagic stroke [139, 140].
Заключение
Nonalcoholic fatty liver disease has become a worldwide heath issue. NASH-associated cirrhosis is the third most common cause of death in NAFLD patients and is predicted to surpass alcoholic liver disease and hepatitis C virus (HCV) as the leading indication for liver transplantation in the U.S. over the next decade. Although NAFLD patients with cirrhosis are at the highest risk of developing hepatocellular carcinoma, we now know that HCC can occur in NAFLD patients in the absence of cirrhosis. Another issue of concern is the increased risk of cardiovascular disease that has been observed in NAFLD patients. Cardiovascular disease is now the leading cause of death in NAFLD patients followed by cancer of the liver. Early detection of patients with nonalcoholic steatohepatitis is of paramount importance if we are to improve patient outcomes through interventional treatment. To that end improvements in diagnostic modalities and drug development are essential.
Литература
- Ludwig J, Viggiano TR, Mcgill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55(7):434–8.
- Fontana RJ, Kleiner DE, Bilonick R, et al. Modeling hepatic fibrosis in African American and Caucasian American patients with chronic hepatitis C virus infection. Hepatology. 2006; 44(4):925–35.
- Caldwell S, Argo C. The natural history of non-alcoholic fatty liver disease. Dig Dis. 2010;28(1):162–8.
- Younossi ZM, Stepanova M, Afendy M, et al. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol Of?cial Clin Pract J Am Gastroenterol Assoc. 2011;9(6):524–30 e521; quiz e560.
- Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. 2013;10(11):627–36.
- Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5:145–71.
- Williams CD, Stengel J, Asike MI, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140 (1):124–31.
- Lazo M, Hernaez R, Eberhardt MS, et al. Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988–1994. Am J Epidemiol. 2013;178(1):38–45.
- Szczepaniak LS, Nurenberg P, Leonard D, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. 2005;288(2):E462–8.
- Patton HM, Yates K, Unalp-Arida A, et al. Association between metabolic syndrome and liver histology among children with nonalcoholic Fatty liver disease. Am J Gastroenterol. 2010; 105(9):2093–102.
- Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated aminotransferase levels in the United States. Am J Gastroenterol. 2003;98(5):960–7.
- Finucane MM, Stevens GA, Cowan MJ, et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet. 2011;377(9765):557–67.
- Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55(6):2005–23.
- Ogden CL, Carroll MD, Flegal KM. Prevalence of obesity in the United States. JAMA. 2014;312(2):189–90.
- Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA. 2014;311(8):806–14.
- Lassailly G, Caiazzo R, Buob D, et al. Bariatric surgery reduces features of nonalcoholic steatohepatitis in morbidly obese patients. Gastroenterology. 2015;149(2):379–88; quiz e315–76.
- Kang H, Greenson JK, Omo JT, et al. Metabolic syndrome is associated with greater histologic severity, higher carbohydrate, and lower fat diet in patients with NAFLD. Am J Gastroenterol. 2006;101(10):2247–53.
- Lee JH, Rhee PL, Lee JK, et al. Role of hyperinsulinemia and glucose intolerance in the pathogenesis of nonalcoholic fatty liver in patients with normal body weight. Korean J Intern Med. 1998;13(1):12–4.
- Marchesini G, Brizi M, Morselli-Labate AM, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. 1999;107(5):450–5.
- Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120(5):1183–92.
- Byrne CD. Ectopic fat, insulin resistance and non-alcoholic fatty liver disease. Proc Nutr Soc. 2013;72(4):412–9.
- Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142(4):711–25 e716.
- Samuel VT, Liu ZX, Qu X, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279(31):32345–53.
- Balent B, Goswami G, Goodloe G, et al. Acute elevation of NEFA causes hyperinsulinemia without effect on insulin secretion rate in healthy human subjects. Ann NY Acad Sci. 2002;967:535– 43.
- Kashyap S, Belfort R, Gastaldelli A, et al. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes. 2003;52(10):2461–74.
- Siddique A, Kowdley KV. Insulin resistance and other metabolic risk factors in the pathogenesis of hepatocellular carcinoma. Clin Liver Dis. 2011;15(2):281–96, vii–x.
- Ekstedt M, Franzen LE, Mathiesen UL, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44(4):865–73.
- Porepa L, Ray JG, Sanchez-Romeu P, Booth GL. Newly diagnosed diabetes mellitus as a risk factor for serious liver disease. CMAJ Can Med Assoc J = J Assoc Med Can. 2010;182 (11):E526–31.
- Harrison SA. Liver disease in patients with diabetes mellitus. J Clin Gastroenterol. 2006;40(1):68–76.
- Leite NC, Salles GF, Araujo AL, Villela-Nogueira CA, Cardoso CR. Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int Of?cial J Int Assoc Study Liver. 2009;29(1):113–9.
- Abdelmalek MF, Liu C, Shuster J, Nelson DR, Asal NR. Familial aggregation of insulin resistance in first-degree relatives of patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol Of?cial Clin Pract J Am Gastroenterol Assoc. 2006;4 (9):1162–9.
- Loomba R, Abraham M, Unalp A, et al. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology. 2012;56(3):943–51.
- Xu L, Ma H, Miao M, Li Y. Impact of subclinical hypothyroidism on the development of non-alcoholic fatty liver disease: a prospective case-control study. J Hepatol. 2012;57(5):1153–4.
- Chung GE, Kim D, Kim W, et al. Non-alcoholic fatty liver disease across the spectrum of hypothyroidism. J Hepatol. 2012;57(1):150–6.
- Adams LA, Feldstein A, Lindor KD, Angulo P. Nonalcoholic fatty liver disease among patients with hypothalamic and pituitary dysfunction. Hepatology. 2004;39(4):909–14.
- Duseja A, Chalasani N. Epidemiology and risk factors of nonalcoholic fatty liver disease (NAFLD). Hepatol Int. 2013;7 (Suppl 2):755–64.
- Hazlehurst JM, Tomlinson JW. Non-alcoholic fatty liver disease in common endocrine disorders. Eur J Endocrinol/Eur Fed Endocr Soc. 2013;169(2):R27–37.
- Newton JL, Jones DE, Henderson E, et al. Fatigue in non-alcoholic fatty liver disease (NAFLD) is significant and associates with inactivity and excessive daytime sleepiness but not with liver disease severity or insulin resistance. Gut. 2008;57 (6):807–13.
- Singh H, Pollock R, Uhanova J, Kryger M, Hawkins K, Minuk GY. Symptoms of obstructive sleep apnea in patients with nonalcoholic fatty liver disease. Dig Dis Sci. 2005;50 (12):2338–43.
- Gambarin-Gelwan M, Kinkhabwala SV, Schiano TD, Bodian C, Yeh HC, Futterweit W. Prevalence of nonalcoholic fatty liver disease in women with polycystic ovary syndrome. Clin Gastroenterol Hepatol Of?cial Clin Pract J Am Gastroenterol Assoc. 2007;5(4):496–501.
- Setji TL, Holland ND, Sanders LL, Pereira KC, Diehl AM, Brown AJ. Nonalcoholic steatohepatitis and nonalcoholic fatty liver disease in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91(5):1741–7.
- Wong VW, Wong GL, Choi PC, et al. Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut. 2010;59(7):969–74.
- Pais R, Charlotte F, Fedchuk L, et al. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J Hepatol. 2013;59(3):550–6.
- Jansen PL. Non-alcoholic steatohepatitis. Eur J Gastroenterol Hepatol. 2004;16(11):1079–85.
- Adams LA, Lymp JF, St Sauver J, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129(1):113–21.
- Nagaoki Y, Hyogo H, Aikata H, et al. Recent trend of clinical features in patients with hepatocellular carcinoma. Hepatol Res Of?cial J Jpn Soc Hepatol. 2012;42(4):368–75.
- Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology. 1990;11(1):74–80.
- Frantzides CT, Carlson MA, Moore RE, et al. Effect of body mass index on nonalcoholic fatty liver disease in patients undergoing minimally invasive bariatric surgery. J Gastrointest Surg Of?cial J Soc Surg Aliment Tract. 2004;8(7):849–55.
- Yatsuji S, Hashimoto E, Tobari M, Taniai M, Tokushige K, Shiratori K. Clinical features and outcomes of cirrhosis due to non-alcoholic steatohepatitis compared with cirrhosis caused by chronic hepatitis C. J Gastroenterol Hepatol. 2009;24(2):248–54.
- Hashimoto E, Yatsuji S, Tobari M, et al. Hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. J Gastroenterol. 2009;44(Suppl 19):89–95.
- Ekstedt M, Hagstrom H, Nasr P, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61(5):1547–54.
- Stefan N, Kantartzis K, Machann J, et al. Identification and characterization of metabolically benign obesity in humans. Arch Intern Med. 2008;168(15):1609–16.
- Lomonaco R, Ortiz-Lopez C, Orsak B, et al. Role of ethnicity in overweight and obese patients with nonalcoholic steatohepatitis. Hepatology. 2011;54(3):837–45.
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121(6):2111–7.
- Halberg N, Wernstedt-Asterholm I, Scherer PE. The adipocyte as an endocrine cell. Endocrinol Metab Clin N Am. 2008;37(3):753–68, x–xi.
- Labonte AC, Tosello-Trampont AC, Hahn YS. The role of macrophage polarization in infectious and inflammatory diseases. Mol Cells. 2014;37(4):275–85.
- Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56(1):16–23.
- Shoelson SE, Lee J, Gold?ne AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–801.
- Weisberg SP, Hunter D, Huber R, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116(1):115–24.
- Nguyen TA, Sanyal AJ. Pathophysiology guided treatment of nonalcoholic steatohepatitis. J Gastroenterol Hepatol. 2012;27 (Suppl 2):58–64.
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–51.
- Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–71.
- Yamaguchi K, Yang L, Mccall S, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45(6):1366–74.
- Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118(9):2992–3002.
- Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 2010;375(9733): 2267–77.
- Leroux A, Ferrere G, Godie V, et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol. 2012;57(1):141–9.
- Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510(7503):84–91.
- Lefterova MI, Lazar MA. New developments in adipogenesis. Trends in endocrinology and metabolism: TEM. 2009;20(3):107–14.
- Yuan M, Konstantopoulos N, Lee J, et al. Reversal of obesityand diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293(5535):1673–7.
- Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291(2):E275–81.
- Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–61.
- Mehal WZ. The Gordian Knot of dysbiosis, obesity and NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10(11):637–44.
- Raman M, Ahmed I, Gillevet PM, et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatology Of?cial Clin Pract J Am Gastroenterol Assoc. 2013;11(7):868–75 e861–63.
- Mouzaki M, Comelli EM, Arendt BM, et al. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. 2013;58(1):120–7.
- Zhu L, Baker SS, Gill C, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57(2):601–9.
- Shanab AA, Scully P, Crosbie O, et al. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig Dis Sci. 2011;56(5):1524–34.
- Wigg AJ, Roberts-Thomson IC, Dymock RB, Mccarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48(2):206–11.
- Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49(6):1877–87.
- Dumas ME, Barton RH, Toye A, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci USA. 2006;103 (33):12511–6.
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444 (7122):1022–3.
- Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–80.
- Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–5.
- Sookoian S, Castano GO, Burgueno AL, Gianotti TF, Rosselli MS, Pirola CJ. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res. 2009;50(10):2111–6.
- Verrijken A, Beckers S, Francque S, et al. A gene variant of PNPLA3, but not of APOC3, is associated with histological parameters of NAFLD in an obese population. Obesity. 2013;21 (10):2138–45.
- Singal AG, Manjunath H, Yopp AC, et al. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta-analysis. Am J Gastroenterol. 2014;109(3):325–34.
- Stickel F, Buch S, Lau K, et al. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology. 2011;53(1):86–95.
- Krawczyk M, Stokes CS, Romeo S, Lammert F. HCC and liver disease risks in homozygous PNPLA3 p. I148M carriers approach monogenic inheritance. J Hepatol. 2015;62(4):980–1.
- He S, Mcphaul C, Li JZ, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285(9):6706–15.
- Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem. 2011;286(43):37085–93.
- Valenti L, Dongiovanni P, Ginanni Corradini S, Burza MA, Romeo S. PNPLA3 I148M variant and hepatocellular carcinoma: a common genetic variant for a rare disease. Dig Liver Dis Of?cial J Ital Soc Gastroenterol Ital Assoc Study Liver. 2013;45 (8):619–24.
- Kozlitina J, Smagris E, Stender S, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46(4):352–6.
- Liu YL, Reeves HL, Burt AD, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. 2014;5:4309.
- Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313(22):2263–73.
- Cheung O, Kapoor A, Puri P, et al. The impact of fat distribution on the severity of nonalcoholic fatty liver disease and metabolic syndrome. Hepatology. 2007;46(4):1091–100.
- Ratziu V, Charlotte F, Heurtier A, et al. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology. 2005;128(7):1898–906.
- Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol Of?cial Clin Pract J Am Gastroenterol Assoc. 2015;13(4):643–54 e641–49; quiz e639–40.
- Hashimoto E, Farrell GC. Will non-invasive markers replace liver biopsy for diagnosing and staging fibrosis in non-alcoholic steatohepatitis? J Gastroenterol Hepatol. 2009;24(4):501–3.
- Maximos M, Bril F, Portillo Sanchez P, et al. The role of liver fat and insulin resistance as determinants of plasma aminotransferase elevation in nonalcoholic fatty liver disease. Hepatology. 2015;61 (1):153–60.
- Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40(6):1387–95.
- Mofrad P, Contos MJ, Haque M, et al. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology. 2003;37(6):1286–92.
- Saadeh S, Younossi ZM, Remer EM, et al. The utility of radiological imaging in nonalcoholic fatty liver disease. Gastroenterology. 2002;123(3):745–50.
- Lin SC, Heba E, Wolfson T, et al. Noninvasive diagnosis of nonalcoholic fatty liver disease and quanti?cation of liver fat using a new quantitative ultrasound technique. Clin Gastroenterol Hepatol Of?cial Clin Pract J Am Gastroenterol Assoc. 2015;13 (7):1337–45 e1336.
- Reeder SB, Cruite I, Hamilton G, Sirlin CB. Quantitative assessment of liver fat with magnetic resonance imaging and spectroscopy. J Magn Reson Imaging JMRI. 2011;34(4), spcone.
- Loomba R, Sirlin CB, Ang B, et al. Ezetimibe for the treatment of nonalcoholic steatohepatitis: assessment by novel magnetic resonance imaging and magnetic resonance elastography in a randomized trial (MOZART trial). Hepatology. 2015;61 (4):1239–50.
- Bastati N, Feier D, Wibmer A, et al. Noninvasive differentiation of simple steatosis and steatohepatitis by using gadoxetic acid-enhanced MR imaging in patients with nonalcoholic fatty liver disease: a proof-of-concept study. Radiology. 2014;271 (3):739–47.
- Musso G. The Finnish Diabetes Risk Score (FINDRISC) and other non-invasive scores for screening of hepatic steatosis and associated cardiometabolic risk. Ann Med. 2011;43(6):413–7.
- Demir M, Lang S, Nierhoff D, et al. Stepwise combination of simple noninvasive fibrosis scoring systems increases diagnostic accuracy in nonalcoholic fatty liver disease. J Clin Gastroenterol. 2013;47(8):719–26.
- Shah AG, Lydecker A, Murray K, Tetri BN, Contos MJ, Sanyal AJ. Comparison of noninvasive markers of fibrosis in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatology Of?cial Clin Pract J Am Gastroenterol Assoc. 2009;7 (10):1104–12.
- Sterling RK, Lissen E, Clumeck N, et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology. 2006;43(6):1317–25.
- Angulo P, Hui JM, Marchesini G, et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology. 2007;45(4):846–54.
- Guha IN, Parkes J, Roderick P, et al. Noninvasive markers of fibrosis in nonalcoholic fatty liver disease: validating the European liver fibrosis panel and exploring simple markers. Hepatology. 2008;47(2):455–60.
- Mcpherson S, Stewart SF, Henderson E, Burt AD, Day CP. Simple non-invasive fibrosis scoring systems can reliably exclude advanced fibrosis in patients with non-alcoholic fatty liver disease. Gut. 2010;59(9):1265–9.
- Ceccarelli S, Panera N, Gnani D, Nobili V. Dual role of microRNAs in NAFLD. Int J Mol Sci. 2013;14(4):8437–55.
- Rottiers V, Naar AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol. 2012;13(4):239–50.
- Lewis AP, Jopling CL. Regulation and biological function of the liver-specific miR-122. Biochem Soc Trans. 2010;38(6):1553–7.
- Esau C, Davis S, Murray SF, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3(2):87–98.
- Iliopoulos D, Drosatos K, Hiyama Y, Goldberg IJ, Zannis VI. MicroRNA-370 controls the expression of microRNA-122 and Cpt1alpha and affects lipid metabolism. J Lipid Res. 2010;51 (6):1513–23.
- Krutzfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438(7068): 685–9.
- Cermelli S, Ruggieri A, Marrero JA, Ioannou GN, Beretta L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE. 2011;6(8):e23937.
- Pirola CJ, Gianotti TF, Castano GO, Sookoian S. Circulating MicroRNA-122 signature in nonalcoholic fatty liver disease and cardiovascular disease: a new endocrine system in metabolic syndrome. Hepatology. 2013;57(6):2545–7.
- Yamada H, Suzuki K, Ichino N, et al. Associations between circulating microRNAs (miR-21, miR-34a, miR-122 and miR-451) and non-alcoholic fatty liver. Clin Chim Acta Int J Clin Chem. 2013;424:99–103.
- Pirola CJ, Fernandez Gianotti T, Castano GO, et al. Circulating microRNA signature in non-alcoholic fatty liver disease: from serum non-coding RNAs to liver histology and disease pathogenesis. Gut. 2015;64(5):800–12.
- Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54(3):603–8.
- Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149(2):367–78 e365; quiz e314–65.
- Kral JG, Thung SN, Biron S, et al. Effects of surgical treatment of the metabolic syndrome on liver fibrosis and cirrhosis. Surgery. 2004;135(1):48–58.
- Dixon JB, Bhathal PS, Hughes NR, O’brien PE. Nonalcoholic fatty liver disease: improvement in liver histological analysis with weight loss. Hepatology. 2004;39(6):1647–54.
- Boden G. Highor low-carbohydrate diets: which is better for weight loss, insulin resistance, and fatty livers? Gastroenterology. 2009;136(5):1490–2.
- Capanni M, Calella F, Biagini MR, et al. Prolonged n-3 polyunsaturated fatty acid supplementation ameliorates hepatic steatosis in patients with non-alcoholic fatty liver disease: a pilot study. Aliment Pharmacol Ther. 2006;23(8):1143–51.
- Assy N, Nassar F, Nasser G, Grosovski M. Olive oil consumption and non-alcoholic fatty liver disease. World J Gastroenterol. 2009;15(15):1809–15.
- Esposito K, Kastorini CM, Panagiotakos DB, Giugliano D. Mediterranean diet and metabolic syndrome: an updated systematic review. Rev Endocr Metab Disord. 2013;14(3):255–63.
- Ryan MC, Itsiopoulos C, Thodis T, et al. The Mediterranean diet improves hepatic steatosis and insulin sensitivity in individuals with non-alcoholic fatty liver disease. J Hepatol. 2013;59(1):138–43.
- Kontogianni MD, Tileli N, Margariti A, et al. Adherence to the Mediterranean diet is associated with the severity of non-alcoholic fatty liver disease. Clin Nutr. 2014;33(4):678–83.
- Kantartzis K, Thamer C, Peter A, et al. High cardiorespiratory ?tness is an independent predictor of the reduction in liver fat during a lifestyle intervention in non-alcoholic fatty liver disease. Gut. 2009;58(9):1281–8.
- Hallsworth K, Fattakhova G, Hollingsworth KG, et al. Resistance exercise reduces liver fat and its mediators in non-alcoholic fatty liver disease independent of weight loss. Gut. 2011;60(9):1278–83.
- Ferrara A, Lewis JD, Quesenberry CP Jr, et al. Cohort study of pioglitazone and cancer incidence in patients with diabetes. Diab Care. 2011;34(4):923–9.
- Lewis JD, Ferrara A, Peng T, et al. Risk of bladder cancer among diabetic patients treated with pioglitazone: interim report of a longitudinal cohort study. Diab Care. 2011;34(4):916–22.
- Soden JS, Devereaux MW, Haas JE, et al. Subcutaneous vitamin E ameliorates liver injury in an in vivo model of steatocholestasis. Hepatology. 2007;46(2):485–95.
- Hoofnagle JH, Van Natta ML, Kleiner DE, et al. Vitamin E and changes in serum alanine aminotransferase levels in patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2013;38 (2):134–43.
- Schurks M, Glynn RJ, Rist PM, Tzourio C, Kurth T. Effects of vitamin E on stroke subtypes: meta-analysis of randomised controlled trials. BMJ. 2010;341:c5702.
- Klein EA, Thompson IM Jr, Tangen CM, et al. Vitamin E and the risk of prostate cancer: the Selenium and vitamin E cancer prevention trial (SELECT). JAMA. 2011;306(14):1549–56.